Tree search with Profile parsimony
Martin R. Smith
2026-02-02
Source:vignettes/profile.Rmd
profile.RmdProfile Parsimony (Faith & Trueman, 2001) finds the tree that is most faithful to the information contained within a given dataset. It is the ‘exact solution’ that implied weights parsimony approximates. For more information on the philosophy and mathematics of profile parsimony, see the companion vignette.
Profile Parsimony is currently implemented in “TreeSearch” for characters with up to two parsimony-informative states. (Further states are treated as ambiguous, whilst retaining as much information as possible.)
Getting started
A companion vignette gives details on installing the package and getting up and running.
Once installed, load the inapplicable package into R using
In order to reproduce the random elements of this document, set a random seed:
# Set a random seed so that random functions in this document are reproducible
RNGversion("3.5.0")## Warning in RNGkind("Mersenne-Twister", "Inversion", "Rounding"): non-uniform
## 'Rounding' sampler used
set.seed(888)Scoring a tree, and conducting a tree search
Here’s an example of using the package to conduct tree search with
profile parsimony. You can load
your own dataset, but for this example, we’ll use a simulated
dataset that comes bundled with the TreeSearch package.
data(congreveLamsdellMatrices)
myMatrix <- congreveLamsdellMatrices[[10]]Unless a starting tree is provided, tree search will from a random addition tree:
additionTree <- AdditionTree(myMatrix, concavity = "profile")
TreeLength(additionTree, myMatrix, "profile")## [1] 552.6187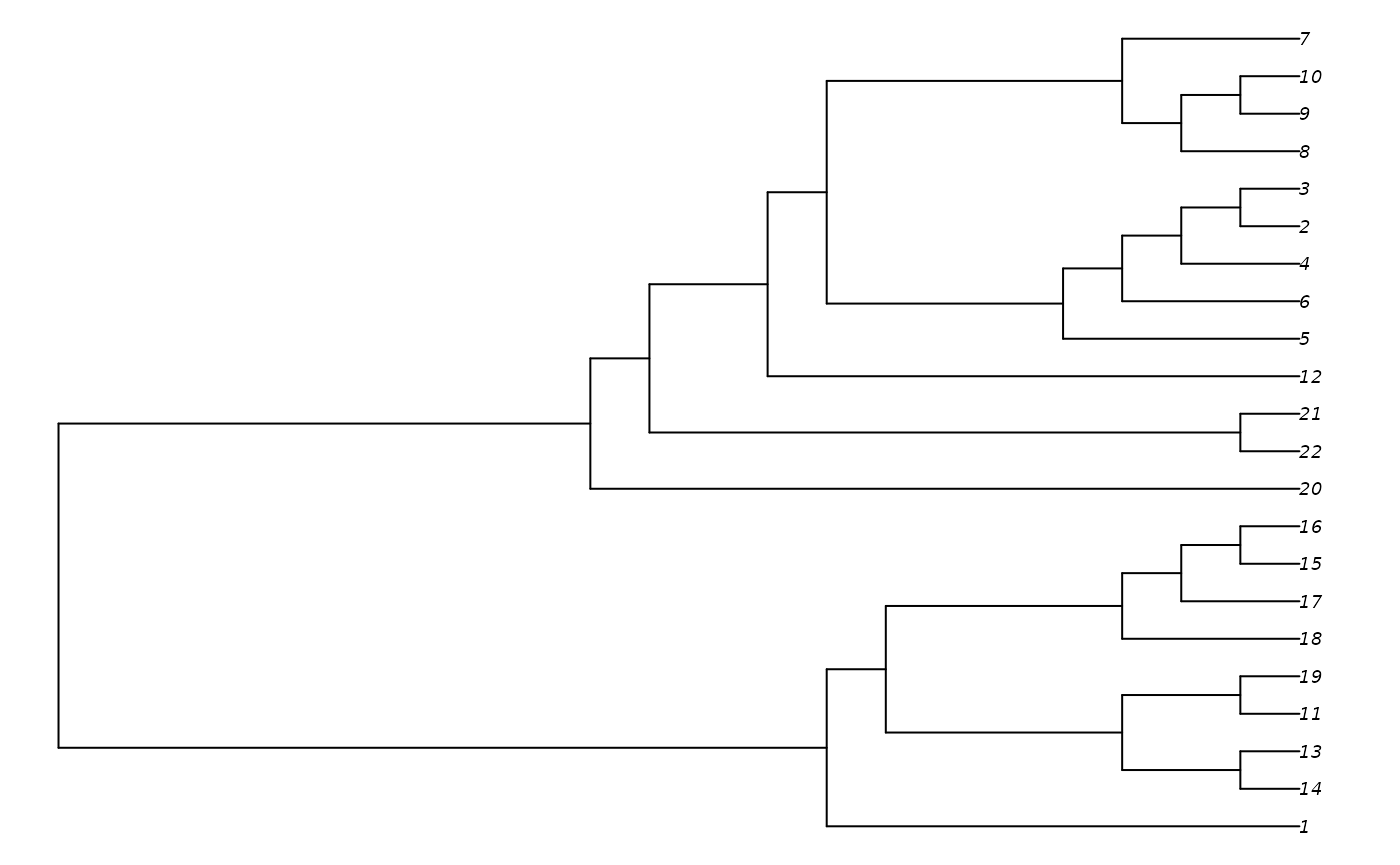
We could alternatively use a random or neighbour-joining tree:
randomTree <- TreeTools::RandomTree(myMatrix, root = TRUE)
TreeLength(randomTree, myMatrix, "profile")## [1] 783.324
njTree <- TreeTools::NJTree(myMatrix)
TreeLength(njTree, myMatrix, "profile")## [1] 540.2259We search for trees with a better score using TBR rearrangements and the parsimony ratchet (Nixon, 1999):
betterTrees <- MaximizeParsimony(myMatrix, additionTree, concavity = "profile",
ratchIter = 3, tbrIter = 3, maxHits = 8)We’ve used very low values of ratchIter,
tbrIter and maxHits for a rapid run, so this
is not necessarily a thorough enough search to find a globally optimal
tree. Nevertheless, let’s see the resultant tree, and its score:
TreeLength(betterTrees[[1]], myMatrix, "profile")## [1] 512.1181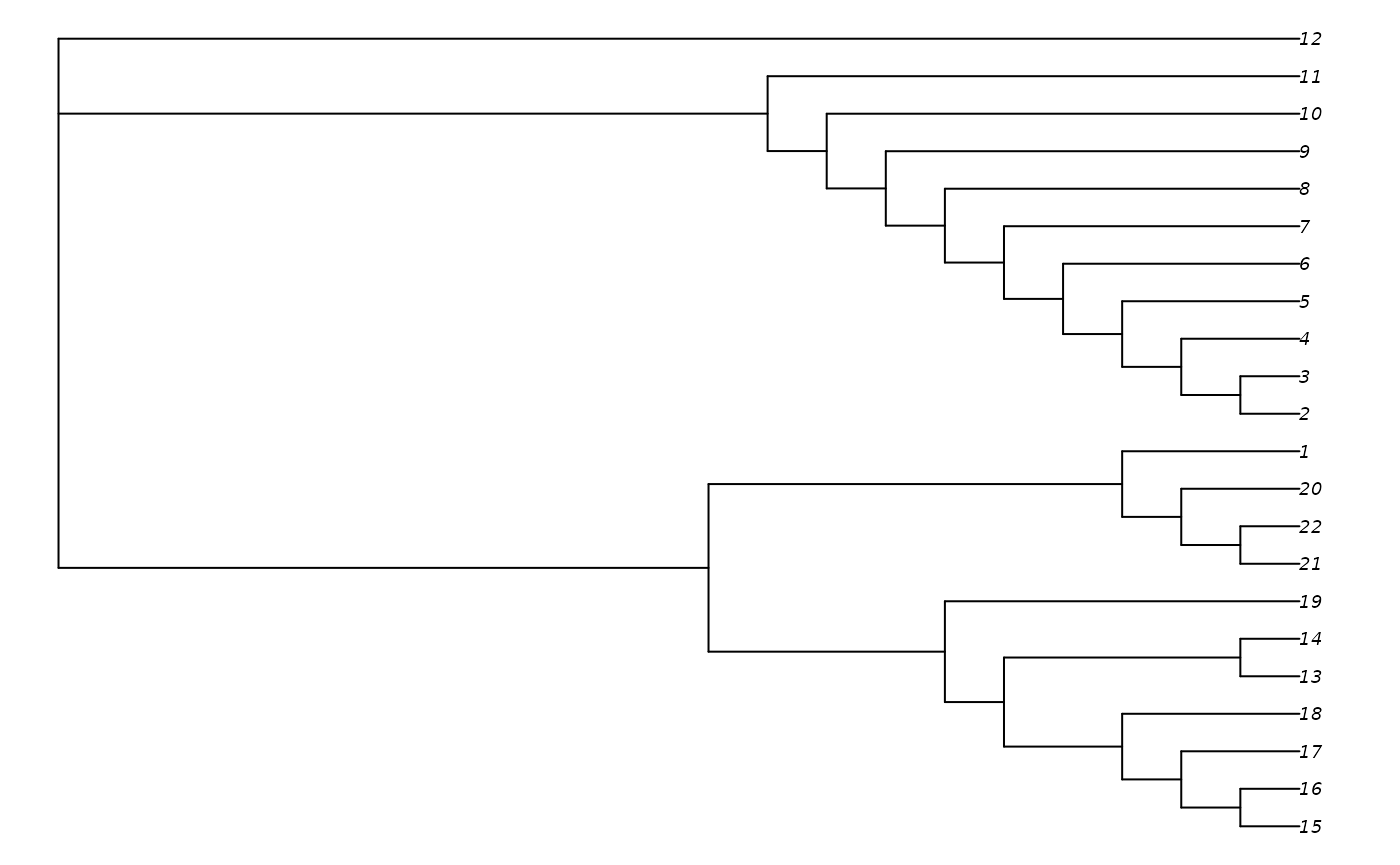
The default parameters may not be enough to find the optimal tree;
type ?MaximizeParsimony to view all search parameters – or
keep repeating the search until tree score stops improving.
View the results
In parsimony search, it is good practice to consider trees that are slightly suboptimal (Smith, 2019).
Here, we’ll take a consensus that includes all trees that are
suboptimal by up to 3 bits. To sample this region of tree space well,
the trick is to use large values of ratchHits and
ratchIter, and small values of searchHits and
searchiter, so that many runs don’t quite hit the optimal
tree. In a serious study, you would want to sample many more than the 3
Ratchet hits (ratchHits) we’ll settle for here, probably
using many more Ratchet iterations.
suboptimals <- MaximizeParsimony(myMatrix, betterTrees, tolerance = 3,
ratchIter = 2, tbrIter = 3,
maxHits = 25,
concavity = "profile")The consensus of these slightly suboptimal trees provides a less resolved, but typically more reliable, summary of the signal with the phylogenetic dataset (Smith, 2019):
par(mar = rep(0.25, 4), cex = 0.75)
table(signif(TreeLength(suboptimals, myMatrix, "profile")))##
## 512.118 513.229 513.897 513.966 514.739 514.849
## 2 1 1 3 1 1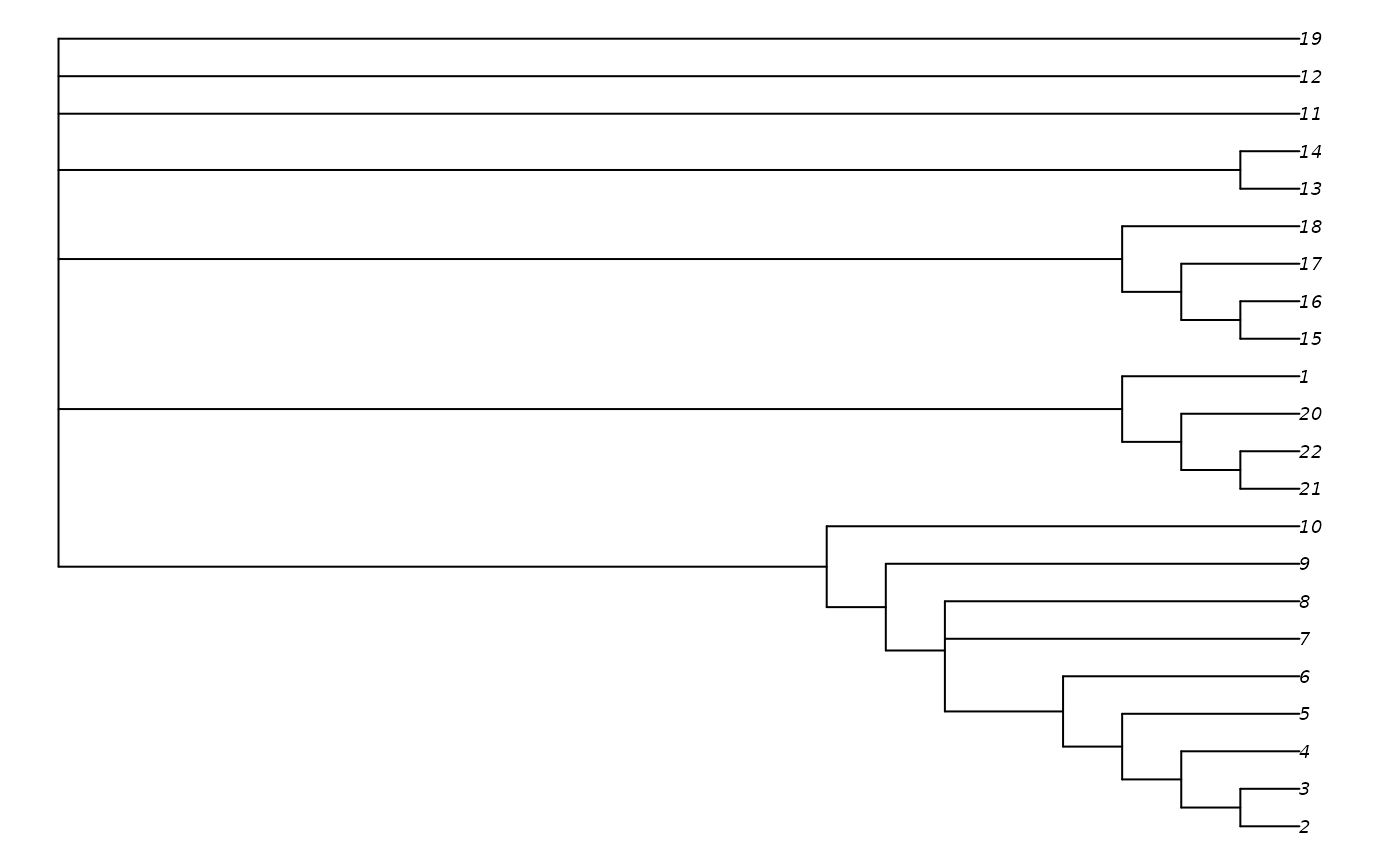
Where next?
-
Search for trees using
- standard parsimony (corrected for inapplicable data)
- custom optimality criteria
Explore the distribution of optimal trees in mappings