3 Corrected parsimony
The phylogenetic dataset contains a considerable proportion of inapplicable codings (1133, = 18.5% of 6108 non-ambiguous tokens; 9.3% of 12150 total cells), which are known to introduce error and bias to phylogenetic reconstruction when the Fitch algorithm is employed (Brazeau et al., 2018; Maddison, 1993). As such, we used the R package TreeSearch v0.1.2 (Smith, 2018) to conduct phylogenetic tree search with a tree-scoring algorithm that avoids logically impossible character transformations when handling inapplicable data (Brazeau et al., 2018), implemented in the MorphyLib C library (Brazeau, Smith, & Guillerme, 2017).
3.1 Search parameters
Heuristic searches were conducted using the parsimony ratchet (Nixon, 1999) under equal and implied weights (Goloboff, 1997). The consensus tree presented in the main manuscript represents a strict consensus of all trees that are most parsimonious under one or more of the concavity constants (k) 3, 4.5, 7, 10.5, 16 and 24, an approach that has been shown to produce higher accuracy (i.e. more nodes and quartets resolved correctly) than equal weights at any given level of precision (Smith, 2017).
3.2 Analysis
The R commands used to conduct the analysis are reproduced below. The results can most readily be replicated using the R markdown files used to generate these pages: in Rstudio, run index.Rmd, then run each block in TreeSearch.Rmd. The complete analysis will take several hours.
3.2.1 Initialize and load data
# Load data from locally downloaded copy of MorphoBank matrix
my_data <- ReadAsPhyDat(nexusFile)
my_data[ignored_taxa] <- NULL
iw_data <- PrepareDataIW(my_data)3.2.2 Generate starting tree
Start by quickly rearranging a neighbour-joining tree, rooted on the outgroup.
nj.tree <- NJTree(my_data)
rooted.tree <- EnforceOutgroup(nj.tree, outgroup)
start.tree <- TreeSearch(tree=rooted.tree, dataset=my_data, maxIter=3000,
EdgeSwapper=RootedNNISwap, verbosity=0)3.2.3 Implied weights analysis
The position of the root does not affect tree score, so we keep it fixed (using RootedXXXSwap functions) to avoid unnecessary swaps.
for (k in kValues) {
iw.tree <- IWRatchet(start.tree, iw_data, concavity=k,
ratchHits = 20, ratchIter=4000, searchHits=56,
swappers=list(RootedTBRSwap, RootedSPRSwap, RootedNNISwap),
verbosity=0L)
score <- IWScore(iw.tree, iw_data, concavity=k)
# Write a single best tree
write.nexus(iw.tree,
file=paste0("TreeSearch/hy_iw_k", k, "_",
signif(score, 5), ".nex", collapse=''))
iw.consensus <- IWRatchetConsensus(iw.tree, iw_data, concavity=k,
swappers=list(RootedTBRSwap, RootedNNISwap),
searchHits=55, searchIter=4000, nSearch=250, verbosity=0L)
write.nexus(iw.consensus,
file=paste0("TreeSearch/hy_iw_k", k, "_",
signif(IWScore(iw.consensus[[1]], iw_data, concavity=k), 5),
".all.nex", collapse=''))
}3.2.4 Equal weights analysis
ew.tree <- Ratchet(start.tree, my_data, verbosity=0L,
ratchHits = 20, ratchIter=4000, searchHits=55,
swappers=list(RootedTBRSwap, RootedSPRSwap, RootedNNISwap))
ew.consensus <- RatchetConsensus(ew.tree, my_data, nSearch=250, searchHits = 85,
swappers=list(RootedTBRSwap, RootedNNISwap),
verbosity=0L)
write.nexus(ew.consensus, file=paste0(collapse='', "TreeSearch/hy_ew_",
Fitch(ew.tree, my_data), ".nex"))3.3 Results
Optimal trees can be downloaded in Nexus format from github.com/ms609/hyoliths/tree/master/TreeSearch.
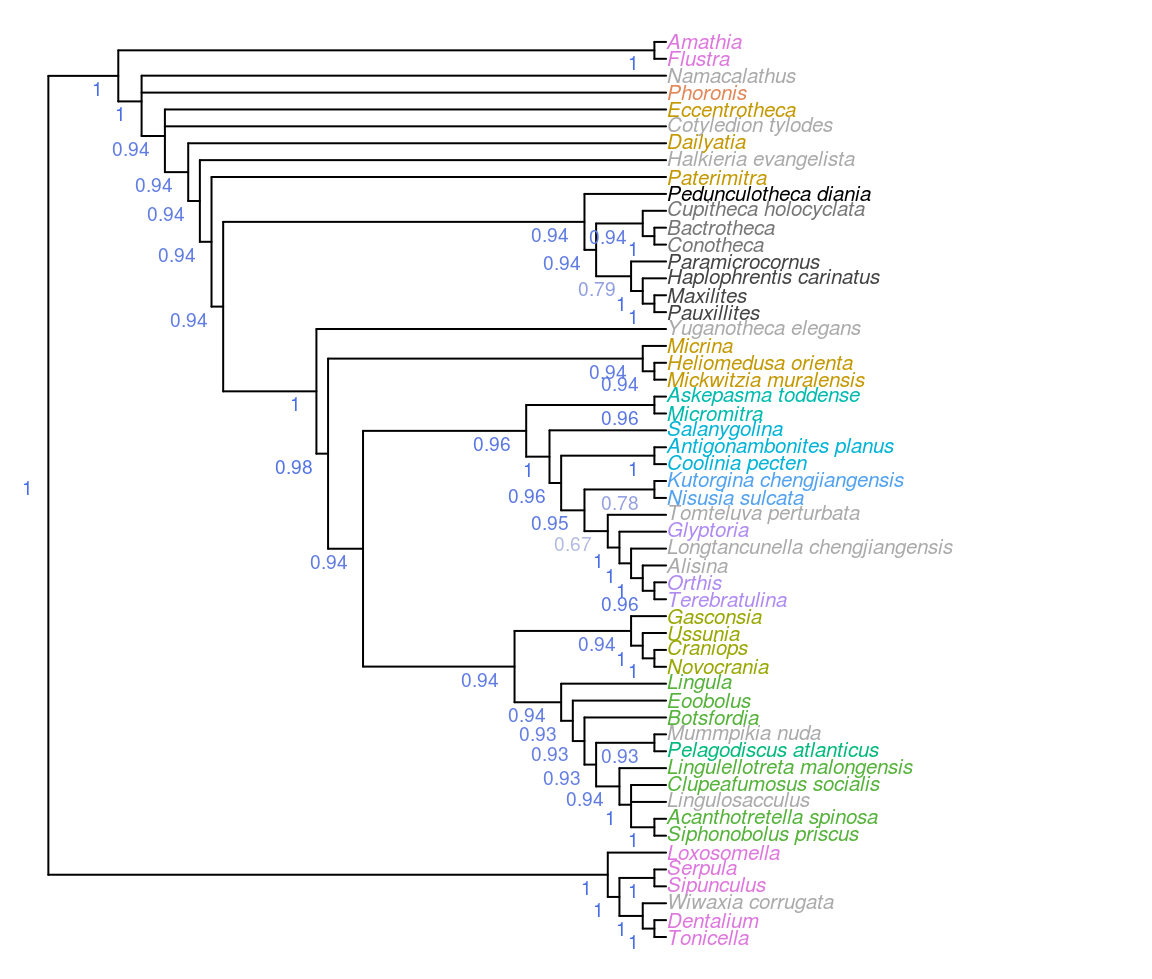
Figure 3.1: Consensus of all parsimony trees, under equal and implied weights. Node labels denote the frequency of each clade in most parsimonious trees under all analytical conditions.
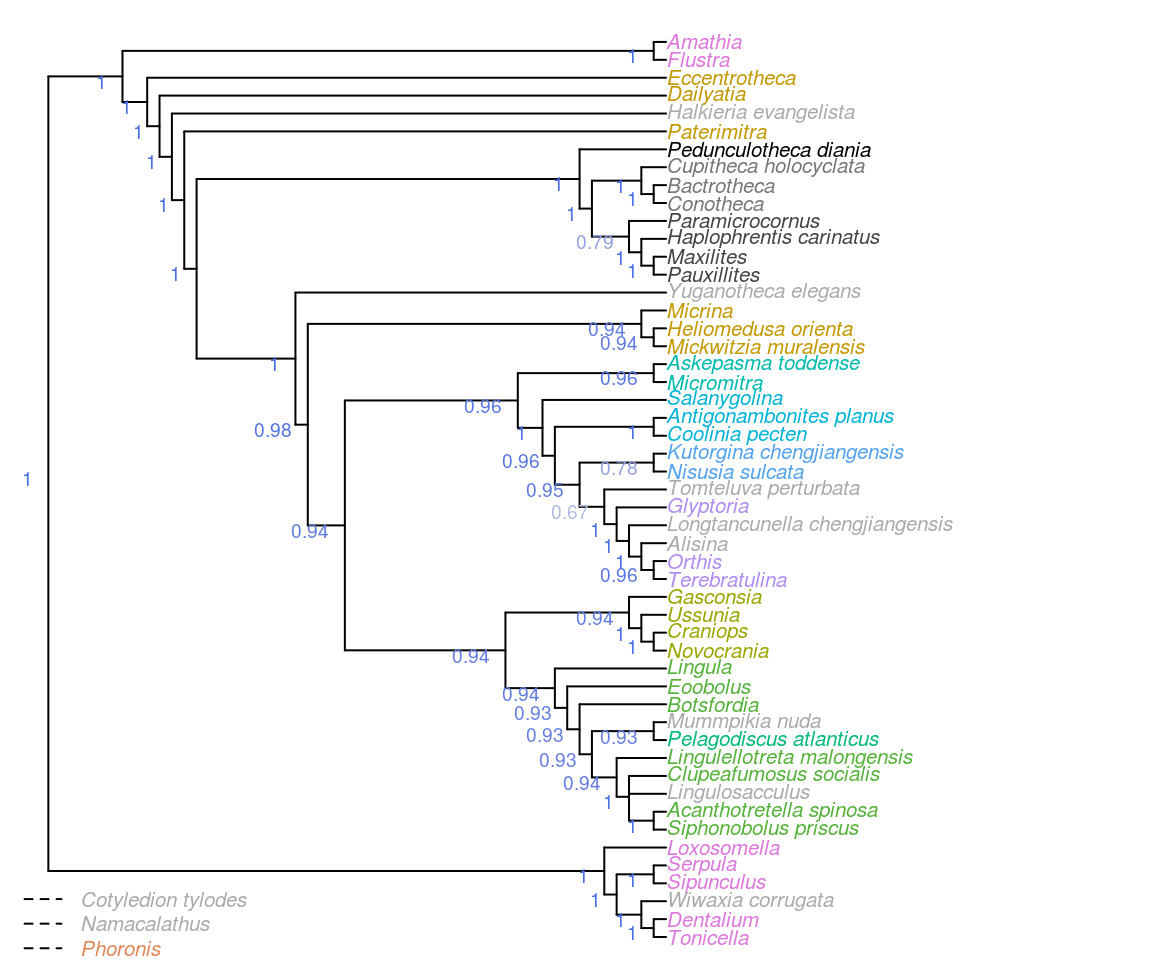
Figure 3.2: Consensus of same trees, with taxa pruned before constructing consensus to give context to clade support. Node labels denote the frequency of each clade in most parsimonious trees under all analytical conditions.
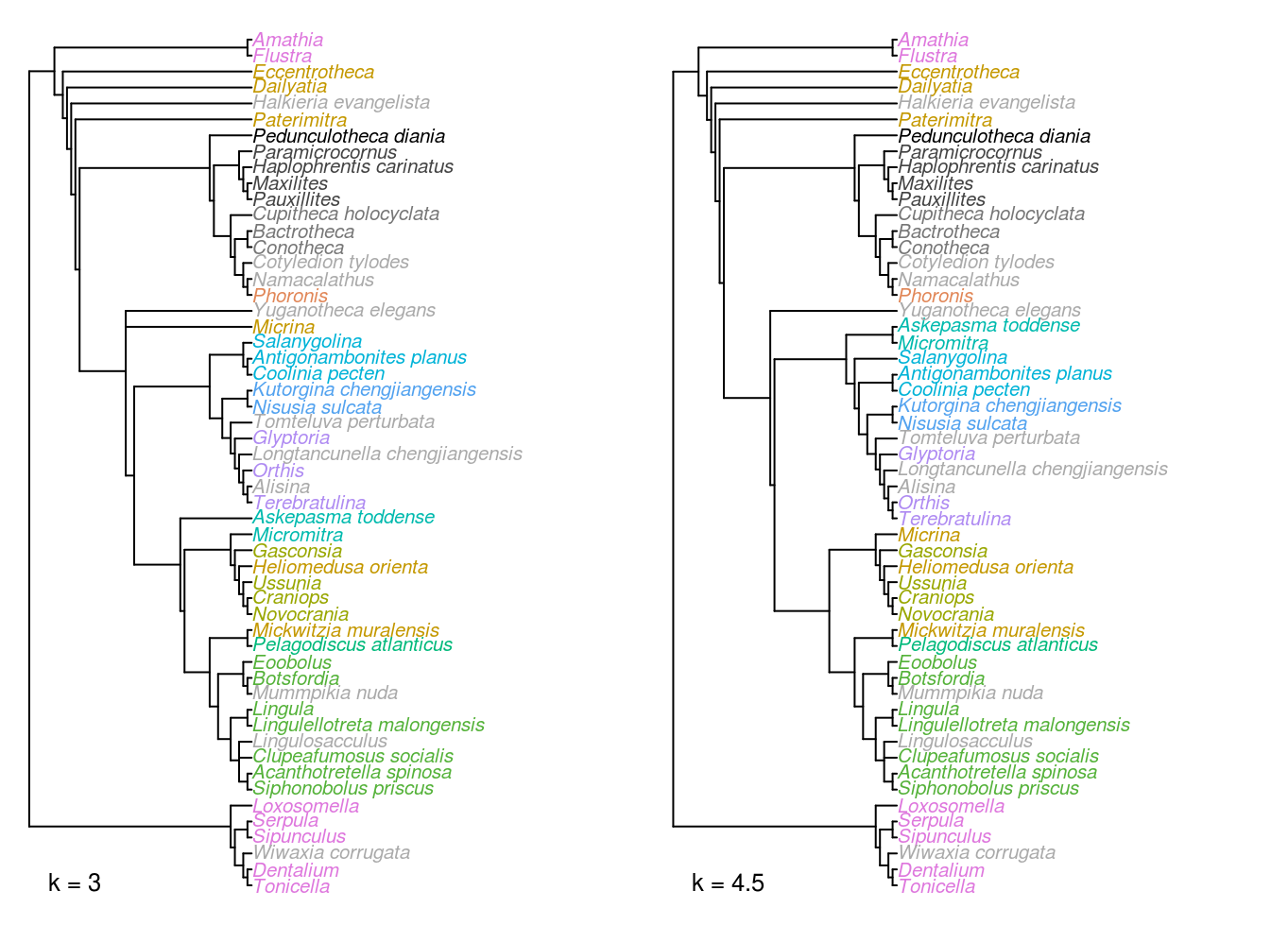
Figure 3.3: Strict consensus trees of implied weights analyses at k = 3 and 4.5.

Figure 3.4: Strict consensus trees of implied weights analyses at k = 7 and 10.5.

Figure 3.5: Strict consensus trees of implied weights analyses at k = 16 and 24.
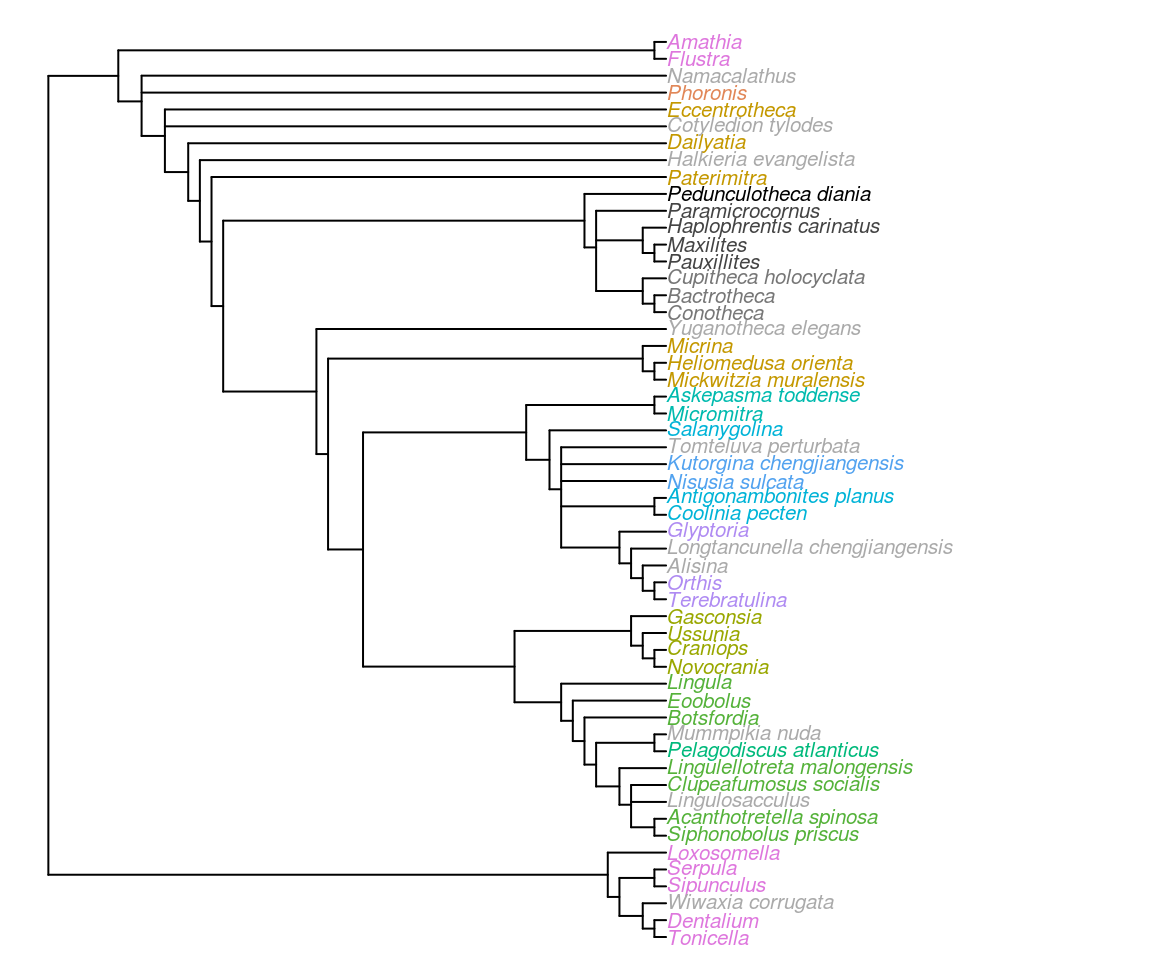
Figure 3.6: Strict consensus of most parsimonious trees under equally weighted parsimony
References
Brazeau, M. D., Guillerme, T., & Smith, M. R. (2018). An algorithm for morphological phylogenetic analysis with inapplicable data. Systematic Biology. doi:10.1101/209775
Maddison, W. P. (1993). Missing data versus missing characters in phylogenetic analysis. Systematic Biology, 42(4), 576–581. doi:10.1093/sysbio/42.4.576
Smith, M. R. (2018). TreeSearch: Phylogenetic tree search using custom optimality criteria. The Comprehensive R Archive Network. doi:10.5281/zenodo.1042590
Brazeau, M. D., Smith, M. R., & Guillerme, T. (2017). MorphyLib: a library for phylogenetic analysis of categorical trait data with inapplicability. Zenodo. doi:10.5281/zenodo.815371
Nixon, K. C. (1999). The Parsimony Ratchet, a new method for rapid parsimony analysis. Cladistics, 15(4), 407–414. doi:10.1111/j.1096-0031.1999.tb00277.x
Goloboff, P. A. (1997). Self-weighted optimization: tree searches and character state reconstructions under implied transformation costs. Cladistics, 13(3), 225–245. doi:10.1111/j.1096-0031.1997.tb00317.x
Smith, M. R. (2017). Quantifying and visualising divergence between pairs of phylogenetic trees: implications for phylogenetic reconstruction. bioR\(\chi\)iv. doi:10.1101/227942