Getting started with Quartet
Martin R. Smith
2026-06-15
Source:vignettes/Using-Quartet.Rmd
Using-Quartet.RmdThis document should contain all you need to get started measuring tree distances with ‘Quartet’. If you get stuck, please let me know so I can improve this documentation.
Loading trees
Instructions for loading phylogenetic trees into R can be found in a separate vignette. For these examples, we’ll enter two simple trees by hand:
Calculating distances
We can calculate distances between pairs of trees using the ‘Quartet’ package.
First we’ll install the package. We can either install the stable version from the CRAN repository:
install.packages("Quartet")or the development version, from GitHub:
devtools::install_github("ms609/Quartet")Then we’ll load the package into the R working environment:
Now the package’s functions are available within R. Let’s proceed to calculate some tree distances.
Pairs of trees
Calculating the distance between two trees is a two stage process. For a quartet distance, we first have to calculate the status of each quartet:
statuses <- QuartetStatus(tree1, tree2)Then we convert these counts into a distance metric (or similarity measure) that suits our needs – perhaps the Quartet Divergence:
QuartetDivergence(statuses, similarity = FALSE)## [1] 0.6031746We can calculate all similarity metrics at once using:
SimilarityMetrics(statuses, similarity = TRUE)## DoNotConflict ExplicitlyAgree StrictJointAssertions
## [1,] 0.3968254 0.3968254 0.3968254
## SemiStrictJointAssertions SymmetricDifference MarczewskiSteinhaus
## [1,] 0.3968254 0.3968254 0.2475248
## SteelPenny QuartetDivergence SimilarityToReference
## [1,] 0.3968254 0.3968254 0.3968254It can be instructive to visualize how each split in the tree is contributing to the quartet similarity:
VisualizeQuartets(tree1, tree2)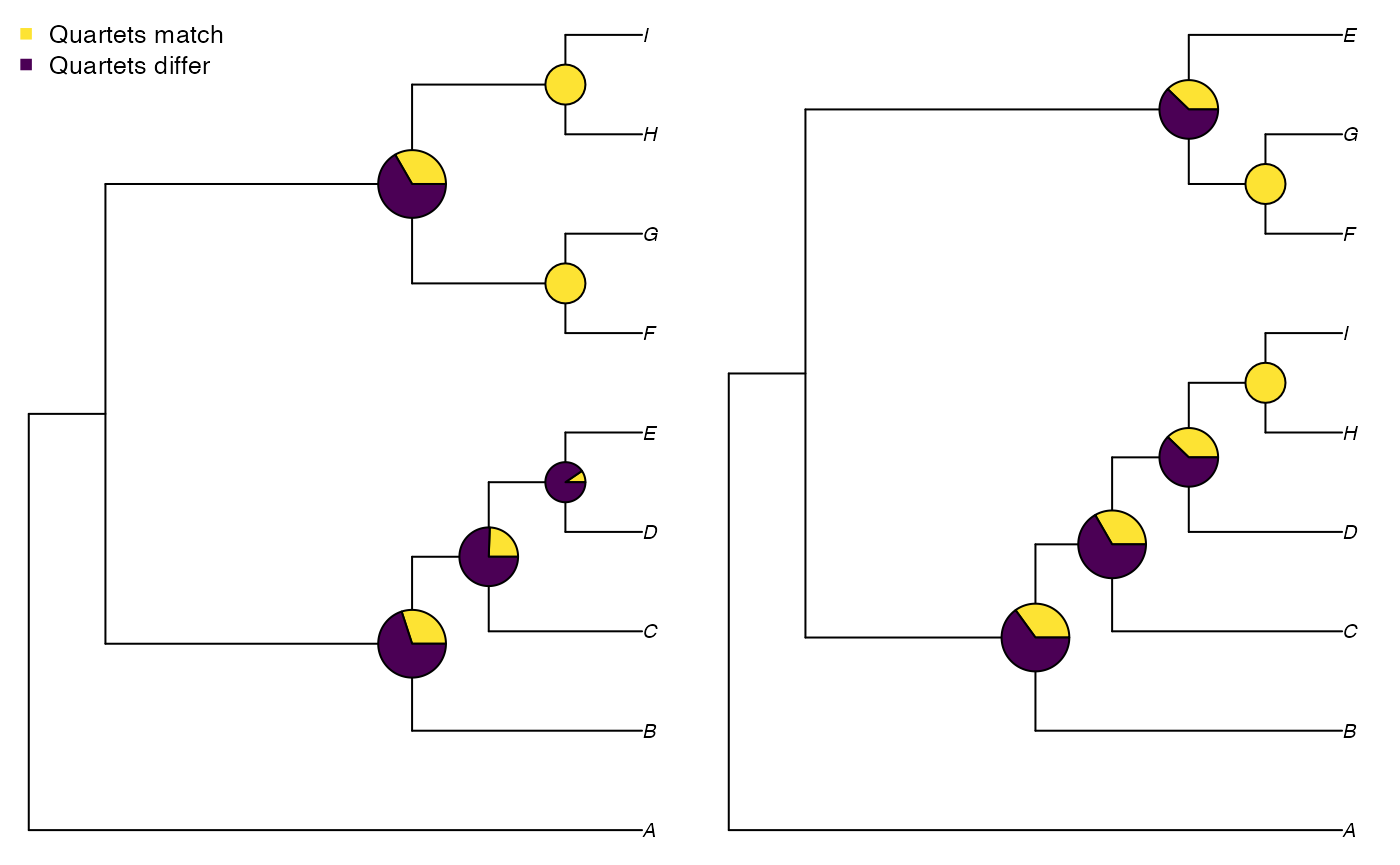
Rather than using quartets, we might want to use partitions as the basis of our comparison:
SimilarityMetrics(SplitStatus(tree1, tree2))## DoNotConflict ExplicitlyAgree StrictJointAssertions
## [1,] 0.3333333 0.3333333 0.3333333
## SemiStrictJointAssertions SymmetricDifference MarczewskiSteinhaus
## [1,] 0.3333333 0.3333333 0.2
## SteelPenny QuartetDivergence SimilarityToReference
## [1,] 0.3333333 0.3333333 0.3333333Multiple comparisons
If you have more than two trees to compare, you can send a list of
trees (class: list or multiPhylo) to the
distance comparison function.
You can calculate the similarity between one tree and a forest of other trees:
library("TreeTools", quietly = TRUE, warn.conflicts = FALSE)
oneTree <- CollapseNode(as.phylo(0, 11), 14)
twoTrees <- structure(list(bal = BalancedTree(11), pec = PectinateTree(11)),
class = "multiPhylo")
status <- SharedQuartetStatus(twoTrees, cf = oneTree)
QuartetDivergence(status)## bal pec
## 0.4939394 0.6272727Or between one tree and (itself and) all other trees in the forest:
forest <- as.phylo(0:5, 11)
names(forest) <- letters[1:6]
status <- SharedQuartetStatus(forest)
QuartetDivergence(status)## a b c d e f
## 1.0000000 0.9757576 0.9757576 0.9333333 0.9121212 0.9333333Or between each pair of trees in a forest:
status <- ManyToManyQuartetAgreement(forest)
QuartetDivergence(status, similarity = FALSE)## a b c d e f
## a 0.00000000 0.02424242 0.02424242 0.06666667 0.08787879 0.06666667
## b 0.02424242 0.00000000 0.02424242 0.08787879 0.06666667 0.06666667
## c 0.02424242 0.02424242 0.00000000 0.08484848 0.08484848 0.04242424
## d 0.06666667 0.08787879 0.08484848 0.00000000 0.04242424 0.04242424
## e 0.08787879 0.06666667 0.08484848 0.04242424 0.00000000 0.04242424
## f 0.06666667 0.06666667 0.04242424 0.04242424 0.04242424 0.00000000Or between one list of trees and a second:
status <- TwoListQuartetAgreement(forest[1:4], forest[5:6])
QuartetDivergence(status, similarity = FALSE)## e f
## a 0.08787879 0.06666667
## b 0.06666667 0.06666667
## c 0.08484848 0.04242424
## d 0.04242424 0.04242424Pairwise comparison
To compute distances between all pairs of trees in a list, use the
PairwiseQuartets() function:
PairwiseQuartets(forest)## a b c d e f
## a 1.0000000 0.9757576 0.9757576 0.9333333 0.9121212 0.9333333
## b 0.9757576 1.0000000 0.9757576 0.9121212 0.9333333 0.9333333
## c 0.9757576 0.9757576 1.0000000 0.9151515 0.9151515 0.9575758
## d 0.9333333 0.9121212 0.9151515 1.0000000 0.9575758 0.9575758
## e 0.9121212 0.9333333 0.9151515 0.9575758 1.0000000 0.9575758
## f 0.9333333 0.9333333 0.9575758 0.9575758 0.9575758 1.0000000
# equivalent to QuartetDivergence(ManyToManyQuartetAgreement(forest))This function can help to summarise sets of trees:
# Map distances between trees
forestDist <- PairwiseQuartets(forest)
mapping <- cmdscale(as.dist(forestDist))
plot(mapping, asp = 1, axes = FALSE, frame.plot = FALSE,
xlab = "", ylab = "", col = seq_along(forest), type = "n")
text(mapping, names(forest))
# The TreeDist library is used to compute the median tree
if (requireNamespace("TreeDist", quietly = TRUE)) {
library("TreeDist")
# Plot the median tree:
plot(median(forest, distance = PairwiseQuartets))
}##
## Attaching package: 'TreeDist'## The following object is masked from 'package:Quartet':
##
## RobinsonFoulds
Trees with different tip labels
“Quartet” can compare trees of different sizes or with non-identical sets of taxa. Quartets pertaining to a leaf that does not occur in one tree are treated as unresolved.
treeAG <- PectinateTree(letters[1:7])
treeBI <- PectinateTree(letters[2:9])
treeEJ <- PectinateTree(letters[5:10])
par(mfrow = c(1, 3), mar = rep(0.3, 4), cex = 1)
plot(treeAG); plot(treeBI); plot(treeEJ)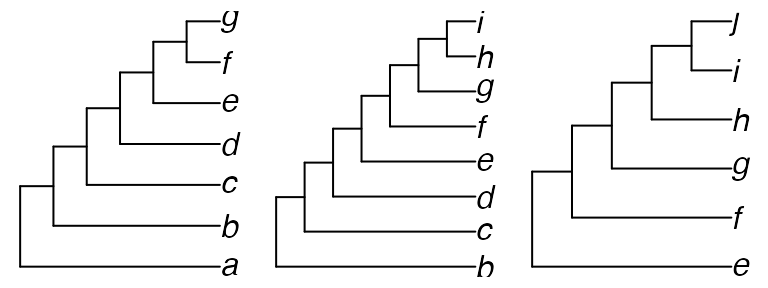
QuartetState(letters[1:4], treeAG) # 3: C is closest to D## [1] 3
QuartetState(letters[1:4], treeBI) # 0: unresolved in this tree## [1] 0
# Calculate status for all leaves observed in trees: here, A..I
QuartetStatus(treeAG, treeBI, nTip = TRUE)## N Q s d r1 r2 u
## [1,] 252 126 15 0 20 55 36
# Calculate status for specified number of leaves
# Here, we have ten taxa A..J, but J does not occur in either of these trees
QuartetStatus(treeAG, treeBI, nTip = 10)## N Q s d r1 r2 u
## [1,] 420 210 15 0 20 55 120
# Compare a list of trees with different numbers of leaves to a reference
QuartetStatus(c(treeAG, treeBI, treeEJ), cf = treeAG, nTip = TRUE)## N Q s d r1 r2 u
## [1,] 420 210 35 0 0 0 175
## [2,] 420 210 15 0 55 20 120
## [3,] 420 210 0 0 15 35 160
# Compare all pairs of trees in a list.
# "u" shows how many possible quartets are unresolved in both trees
ManyToManyQuartetAgreement(c(treeAG, treeBI, treeEJ), nTip = TRUE)[, , "u"]## [,1] [,2] [,3]
## [1,] 175 120 160
## [2,] 120 140 130
## [3,] 160 130 195Other calculations
To calculate how many quartets are unique to a certain tree (akin to
the partitionwise equivalent ape::prop.clades), use:
interestingTree <- as.phylo(42, 7)
referenceTrees <- list(BalancedTree(7), PectinateTree(7))
status <- CompareQuartetsMulti(interestingTree, referenceTrees)status["x_only"] = 23 quartets are resolved in a certain
way in interestingTree, but not resolved that way in any
referenceTrees.
What next?
You may wish to:
Read more about Quartet distances
Review alternative distance measures and corresponding functions
Interpret or contextualize tree distance metrics